
DNA Contamination of RNA Vaccines

Pieces of DNA in two batches of Pfizer vaccine. The vial content was analyzed.
the pieces of DNA are small and are likely to damage the human genome by integrating and
becoming permanent mutations (like shotgun pellets hitting a washboard). its important to look at
DNA taken from different body tissues of vaccinated people to see if this is happening and if it can
be causing any adverse events now or if there is a future cancer risk down the road. we should
sequence a few hundred people and find out if this DNA ever got into the human genome.
we used the sequences of all the little pieces of DNA in the vaccine to reconstruct the actual sequence of where it came from. It is the plasmid used in production of the mRNA (pBK-CMV modified to contain SPIKE gene). The DNA in the vaccine is a contaminate leftover from the process used in large scale production. This DNA was not present in the material used in the trials because the process of making the stuff was different (it did not use this plasmid DNA).
We have a pretty easy and cheap method to detect one of the pieces of plasmid DNA. It’s a PCR test similar to what we used to detect SARS-COV2 during the pandemic (the saliva test).
There are about 2 billion copies of the fragment containing the origin of replication, and from nanopore sequence analysis, there is probably 50-100 times that many pieces of plasmid DNA derived from the entire vector.
This means each shot has about 200 billion pieces of plasmid DNA encapsulated in the lipid nanoparticle.
This is a bad idea.
Conclusions
We should check a bunch of vaccinated people to see if plasmid DNA has integrated into their genomic DNA. We (you) should insist that the USFDA force Pfizer to get the DNA out of the booster and all future mRNA based vaccines.
The regulation that allowed the DNA to be there should be changed. It’s a leftover from previous vaccines that contained only naked DNA. The mRNA vaccines have this DNA encapsulated in a lipid nanoparticle delivery system (trojan horse) and so the DNA is a far more serious issue. 20 Greek soldiers wandering around outside the walls of Troy are not a big deal.
20 Greek soldiers packed inside a large wooden horse are a different matter.**
Philips Buckhaults, Ph.D. Professor of Cancer Molecular Genetics University of South Carolina
Sequencing of bivalent Moderna and Pfizer mRNA vaccines reveals nanogram to microgram quantities of expression vector dsDNA per dose
Kevin McKernan, Yvonne Helbert, Liam T. Kane, Stephen McLaughlin Medicinal Genomics, 100 Cummings Center, Suite 406-L, Beverly Mass, 01915
Several methods were deployed to assess the nucleic acid composition of four vials of the Moderna and Pfizer bivalent mRNA vaccines. Two vials from each vendor were evaluated with Illumina sequencing, qPCR, RT-qPCR, Qubit™ 3 fluorometry and Agilent Tape Station™ electrophoresis. Multiple assays support DNA contamination that exceeds the European Medicines Agency (EMA) 330ng/mg requirement and the FDAs 10ng/dose requirements. These data may impact the surveillance of vaccine mRNA in breast milk or plasma as RT-qPCR assays targeting the vaccine mRNA cannot discern DNA from RNA without RNase or DNase nuclease treatments. Likewise, studies evaluating the reverse transcriptase activity of LINE-1 and vaccine mRNA will need to account for the high levels of DNA contamination in the vaccines. The exact ratio of linear fragmented DNA versus intact circular plasmid DNA is still being investigated. Quantitative PCR assays used to track the DNA contamination are described.
Introduction
Several studies have made note of prolonged presence of vaccine mRNA in breast milk and plasma (Bansal et al. 2021; Hanna et al. 2022; Castruita et al. 2023). This could be the result of the stability of N1-methylpseudouridine (m1Ψ) in the mRNA of the vaccine. Nance et al. depict a vaccine mRNA synthesis method that utilizes a dsDNA plasmid that is first amplified in E.coli prior to an in-vitro T7 polymerase synthesis of vaccine mRNA (Nance and Meier 2021). Failure to remove this DNA could result in the injection of spike encoded nucleic acids more stable than the modified RNA. The EMA has stated limits at 330ng/mg of DNA to RNA (Josephson 2020-11-19). The FDA has issued guidance for under 10ng/dose in vaccines (Sheng-Fowler et al. 2009).
Residual injected DNA can result in type I interferon responses and can increase the potential for DNA integration(Ulrich-Lewis et al. 2022).
Results
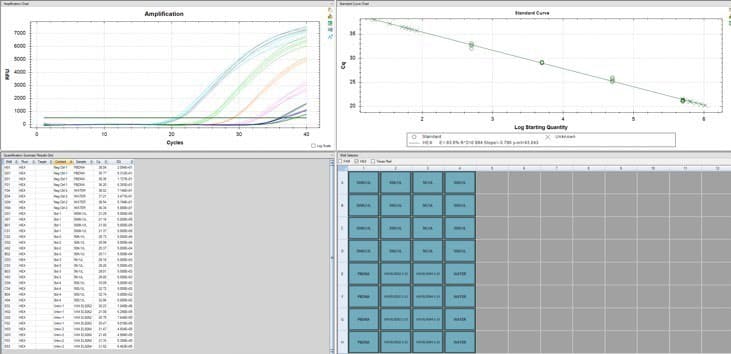
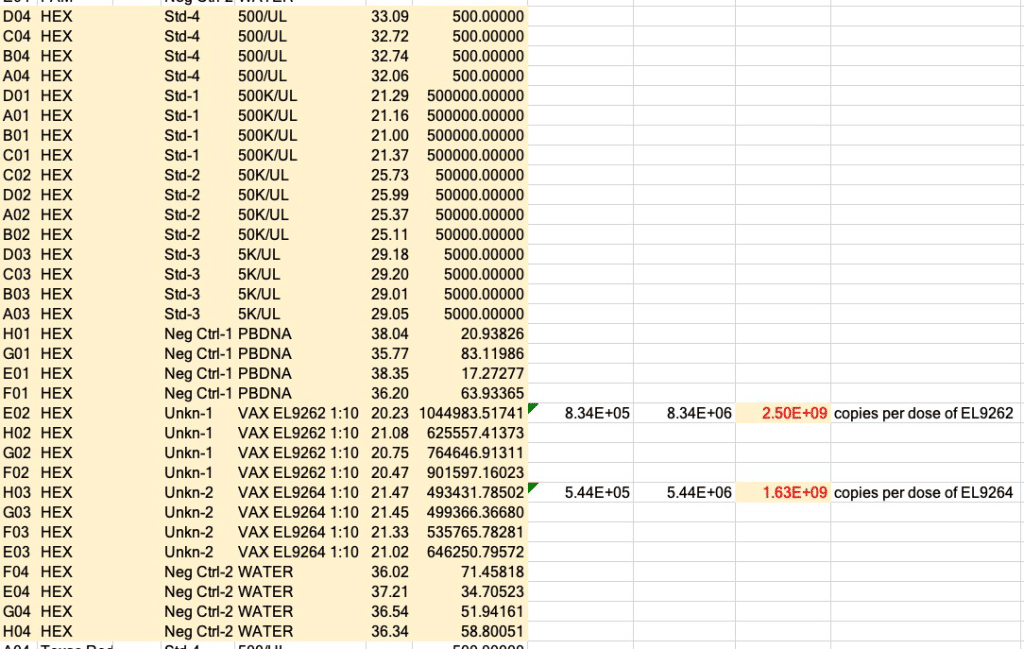
To assess the nucleic acid composition of the vaccines, vaccine DNA was deeply sequenced using two different methods. The first method used a commercially available New England Biolabs RNA-seq method that favored the sequencing of the RNA but still presented over 500X coverage for the unanticipated DNA vectors (Figure 1 and 2). The RNA-seq assemblies had truncated poly A tracts compared to the constructs described by Nance et al. The second method eliminated the RNA with RNase A treatment and sequenced only the DNA using a Watchmaker Genomics fragment library kit. The DNA focused assemblies delivered vector assemblies with more intact poly A tracts (Figure 3).
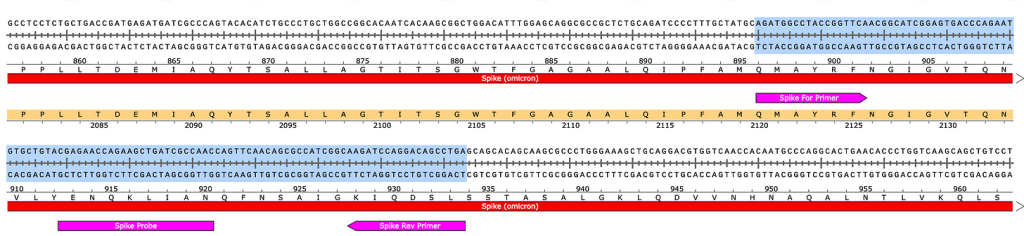
These assemblies were utilized to design multiplex qPCR and RT-qPCR assays that target the spike sequence present in both the vaccine mRNA and the DNA vector while also targeting then origin of replication sequence present only in the DNA vector (Figure 3). The assembly of Pfizer vial 1 contains a 72bp insertion not present in the assembly of Pfizer vial 2. This indel is known for its enhancement to the SV40 promoter and its nuclear targeting signal (Dean et al. 1999) (Moreau et al. 1981).
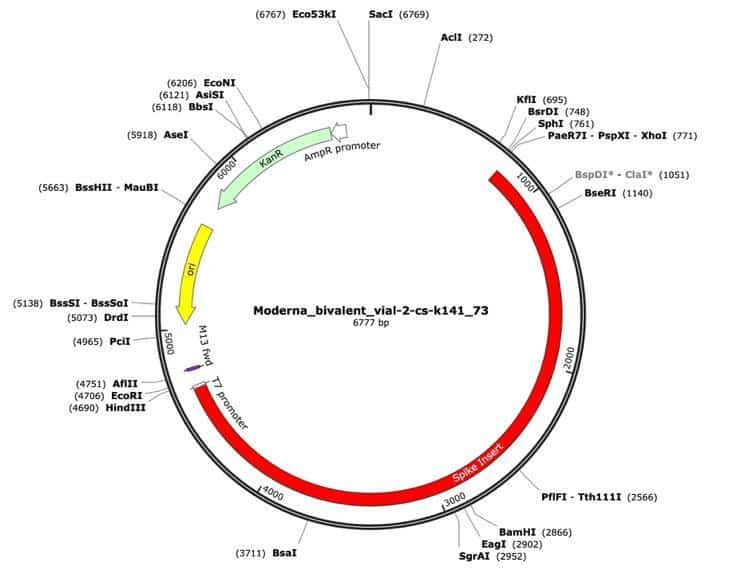
Figure 1. A Moderna vector assembly of an RNA-seq library with a spike insert (red), Kanamycin resistance gene (green) driven by an AmpR promoter and a high copy bacterial origin of replication (yellow).
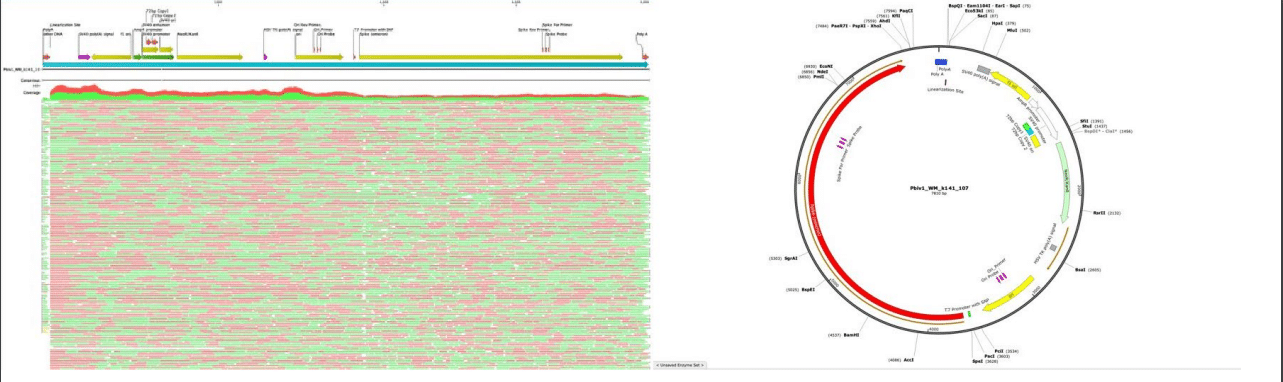
Figure 2. Pfizer bivalent vaccine assembly of the RNA-seq library. Annotated with SEB/FCS, spike insert (red), bacterial origin of replication (yellow), Neo/Kan resistance gene(green), F1 origin (yellow) and an SV40 promoter (yellow and white).
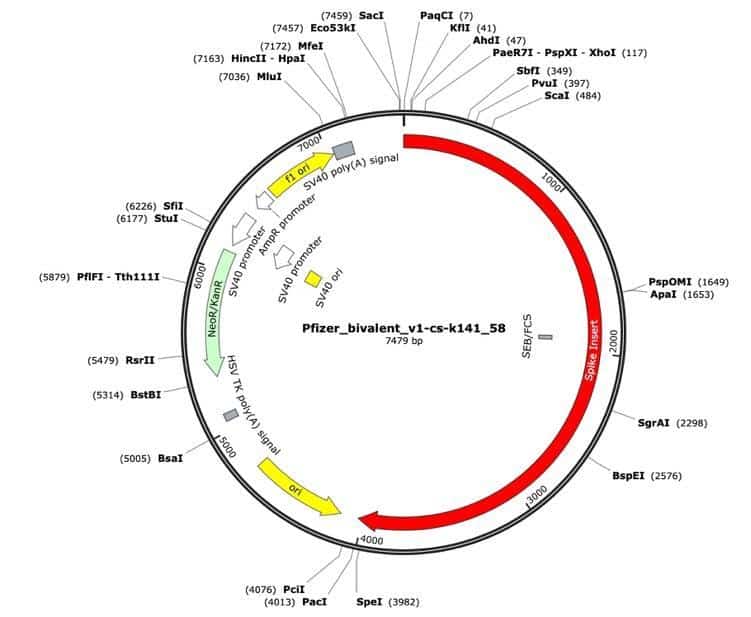
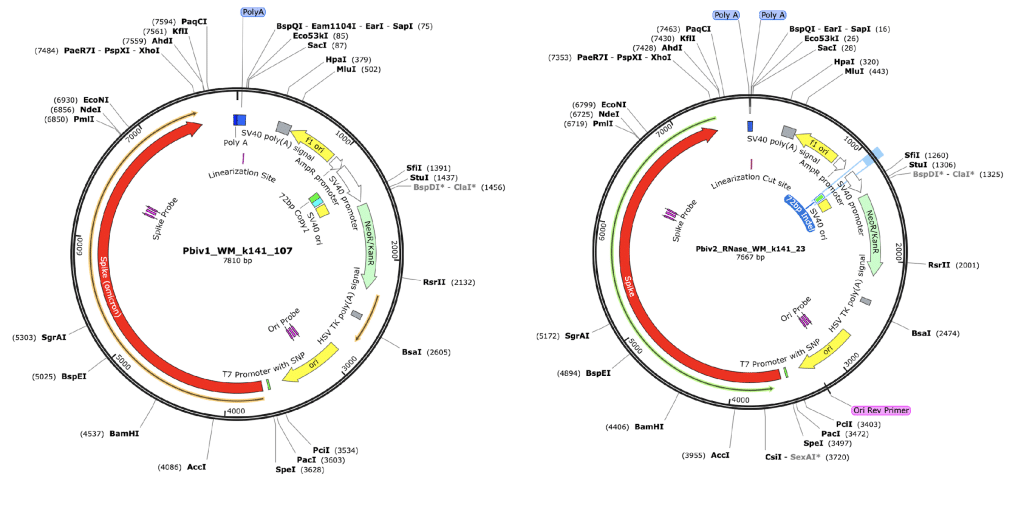
Figure 3. RNase treated vaccines were shotgun sequenced with Illumina (RNase-Seq not RNA-seq). Pfizer vectors from vial 1 (left) and vial 2 (right) contain a 72bp difference in the SV40 promoter (green and light blue annotation). qPCR assays are depicted in pink as Spike probe and Ori probe. The RNase sequencing provided better resolution over the Eam1104i linearization site and the Poly adenylation sequence. The vectors differ in the length of the polyA tail (likely sequencing artifact) and the 72bp indel.
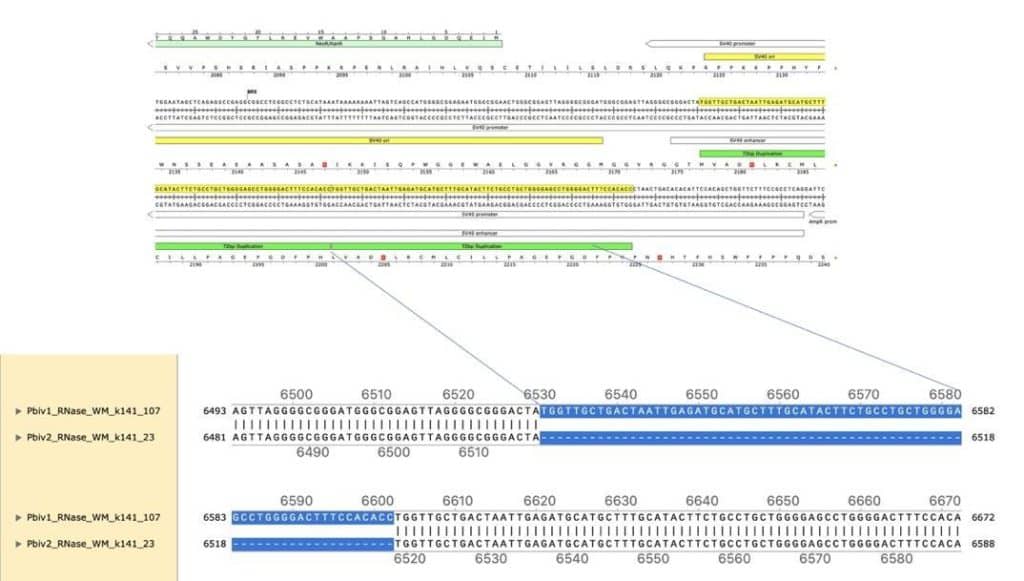
Figure 4. Local alignment of Pfizer vial 1 to Pfizer vial 2 vectors highlights the 72bp tandem duplication in blue.
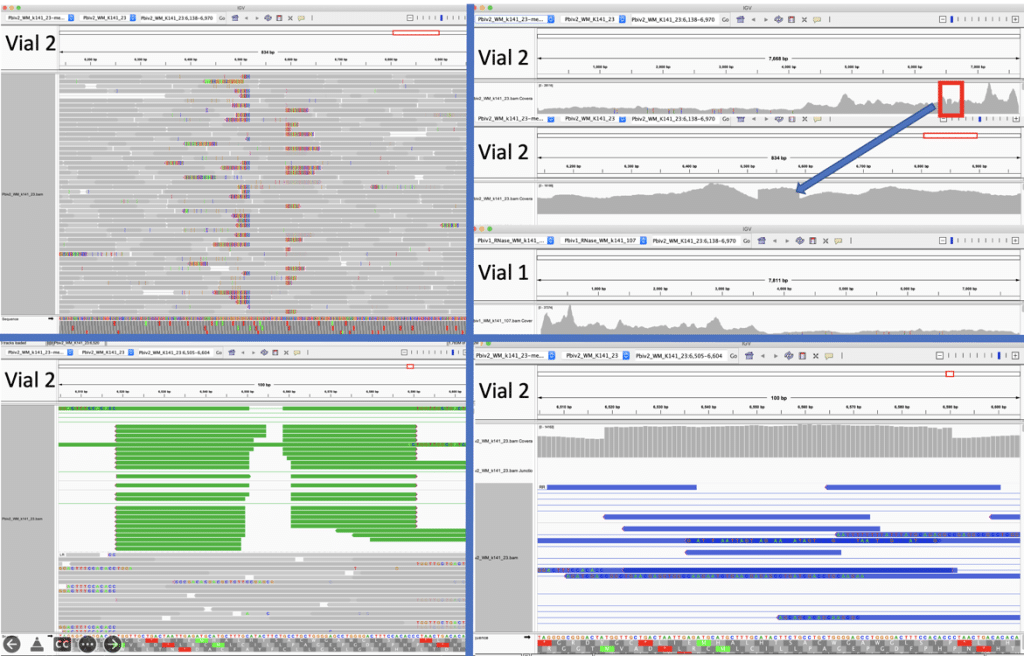
Figure 5A. Close inspection of the Integrative Genome Viewer (IGV) demonstrates the appearance of a 72bp insertion that is heteroplasmic in Pfizer vial 2. The upper left IGV view is a zoomed-out view where the colored marks depict the indel. The lower Left IGV view shows inverted paired reads as the 72bp insertion is a tandem repeat and paired reads shorter than 72bp can be mapped two different ways. Upper Right IGV view demonstrates a read coverage pile up or ‘Plateau’. This occurs when the reference has one copy of the 72bp repeat and the sample has 2 copies. Note- In the upper right IGV depiction, the sequence in Vial 1 is in the opposite orientation in IGV as Vial 2. Lower right IGV view is a zoomed view of the upper right IGV screen.
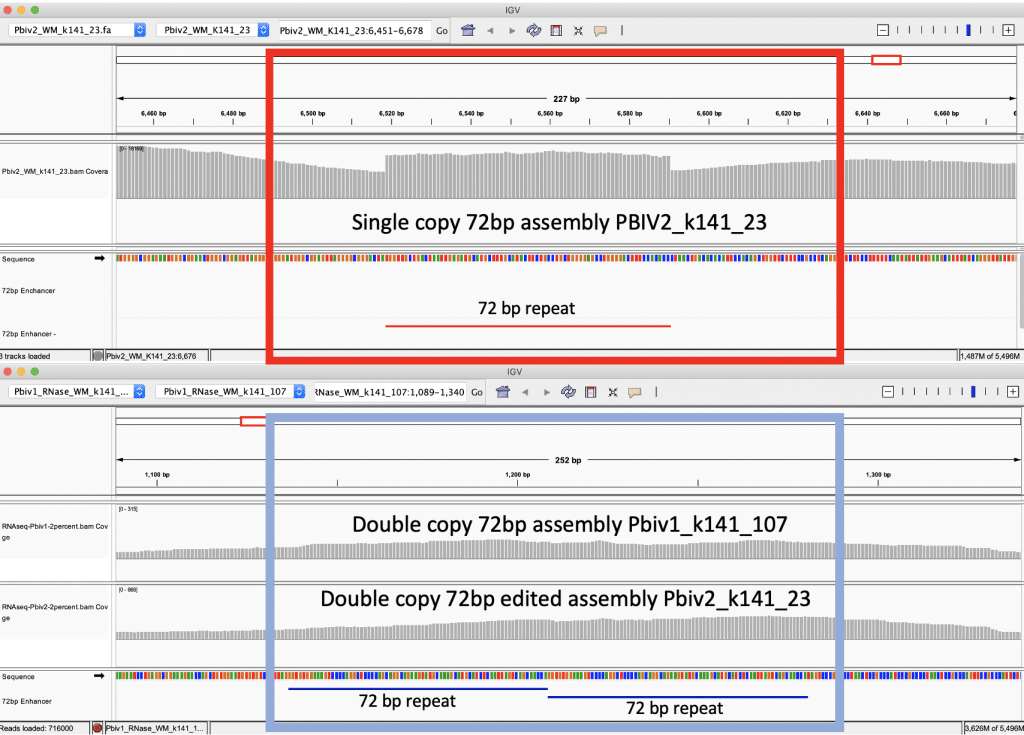
Figure 5B. IGV view of the read coverage over Pbiv2_k141_23 shows a discrete 72bp plateau in coverage (red rectangle). Editing the Pbiv2_k141_23 reference to include 2 copies of the 72bp sequence, and remapping the sequence data to this corrected sequence shows that the coverage over both vectors is more normal with no coverage plateau in Pfizer vial 2.
These data conclude that all Pfizer vectors contain a homoplastic 2 copy 72bp SV40 Enhancer associated with more robust expression and nuclear localization. The initial heteroplastic indel was an artifact of the Megahit assembler and short insert libraries. These vectors contain an SV40 Promoter, SV40 Enhancer, SV40 Origin, and an SV40 polyA signal. They do not contain the entire SV40 virus or the SV40 T-antigen.
To estimate the size of the DNA, the purified vaccines were evaluated on an Agilent Tape Station™ using DNA (genomic DNA screen tapes) and RNA based (high sensitivity RNA tapes) electrophoresis tapes.
Agilent Tape Station™ electrophoresis reveal 7.5 – 11.3 ng/µl of dsDNA compared to the 23.7 -55.9ng/µl of mRNA detected in each 300µl sample. Qubit™ 3 fluorometry estimated 1-2.8ng/µl of DNA and 21.8ng – 52.8ng/µl of RNA. There is higher fragmentation seen in the DNA electrophoresis. The total RNA levels are less than the anticipated 30ug (100ng/µl) and 100ug (200ng/µl) doses suggesting a loss of yield in DNA and RNA isolation, manufacturing variance or RNA decay with questionable cold chains.
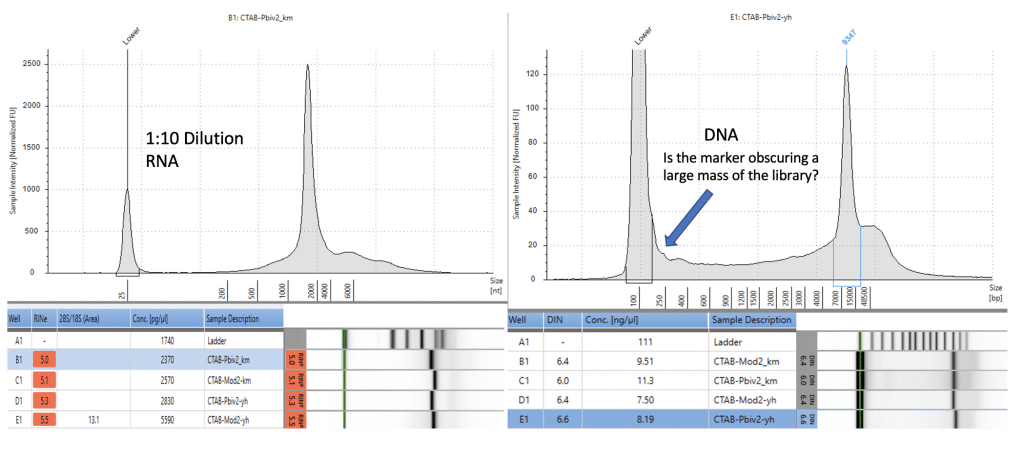
Figure 6. Agilent Tape Station™ electrophoresis demonstrates 23.7ng/µl – 55.9ng/µl of RNA (left). 7.5ng-11.3ng/µl are observed on DNA based Tape Station™. While the DNA electropherogram shows a peak suggestive of a full-length plasmid, this sample is known to have high amounts of N1-methylpseudouridine RNA present. DNA hybrids with N1-methylpseudouridine mRNA may provide enough intercalating dye cross talk to produce a peak. The sizing of the peak on the RNA tape on the left is shorter than expected. This may be the results of N1 methylpseudouridine changing the secondary structure or the mass to charge ratio of the DNA.
Quantitative PCR assays were designed using IDTs Primer Quest software targeting a region in the spike protein that was identical between Moderna and Pfizer spike sequences and a shared sequence in the vectors’ origin of replication. This allowed the qPCR and RT-qPCR assessment of the vaccines. qPCR only amplifies DNA while RT-qPCR amplifies both DNA and RNA. Gradient qPCR was utilized to explore conditions where both targets would perform under the same cycling conditions for both RT-qPCR and PCR (gradient PCR data not shown).
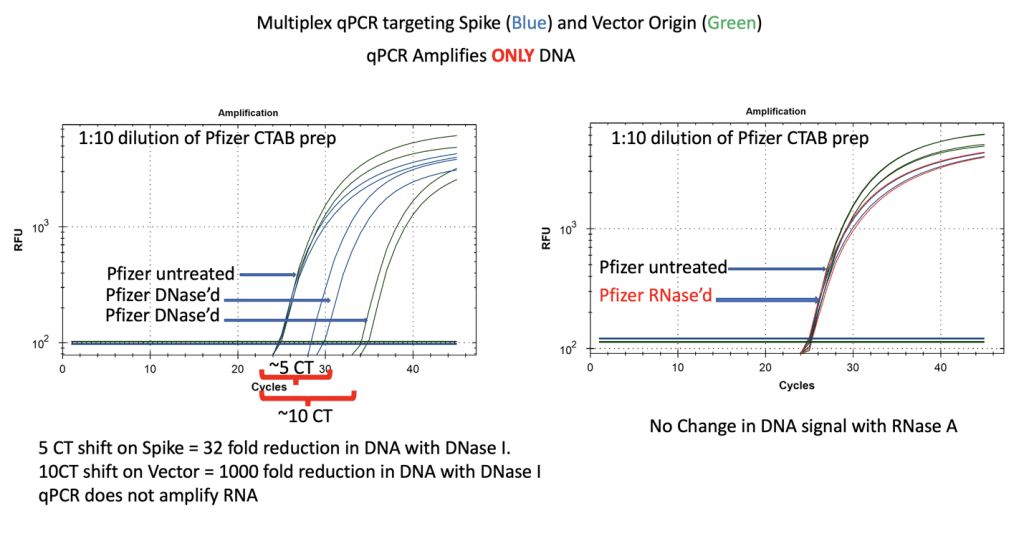
Figure 7. qPCR of Pfizer’s bivalent vaccine with and without DNase I (left) and RNase A (right). Untreated mRNA demonstrates equal CTs for Spike and Vector assays as expected. Vector is more DNase I sensitive than the Spike suggesting the modRNA may inhibit nuclease activity of DNase I against complementary DNA targets. RNase A treatment doesn’t alter the qPCR signal. Non Template Control (NTC) amplification produces no signal out to CT 40 with the spike assay and no signal out to CT 37 with the bacterial origin of replication assay. This background Ori CT may vary with different polymerases that are expressed in bacteria vectors containing this common Ori.
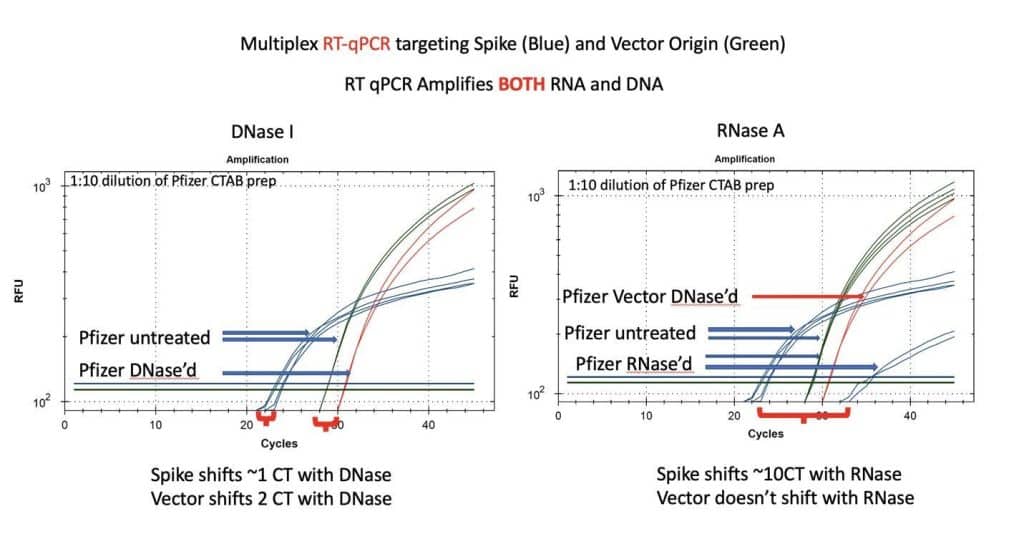
Figure 8. RT-qPCR amplifies both DNA and RNA. The untreated samples show a large CT offset with Pfizer Spike and Vector assays (Left Blue versus Green). This is anticipated as the T7 polymerization should create more mRNA over spike than over the vector. Small 1-2 CT shifts are seen with DNase I treatment. This is expected if the DNA is less than equal concentration of nucleic acid in RT-PCR. RNase treatment (Right) shows a 10 CT offset but doesn’t alter the DNA vector CT.
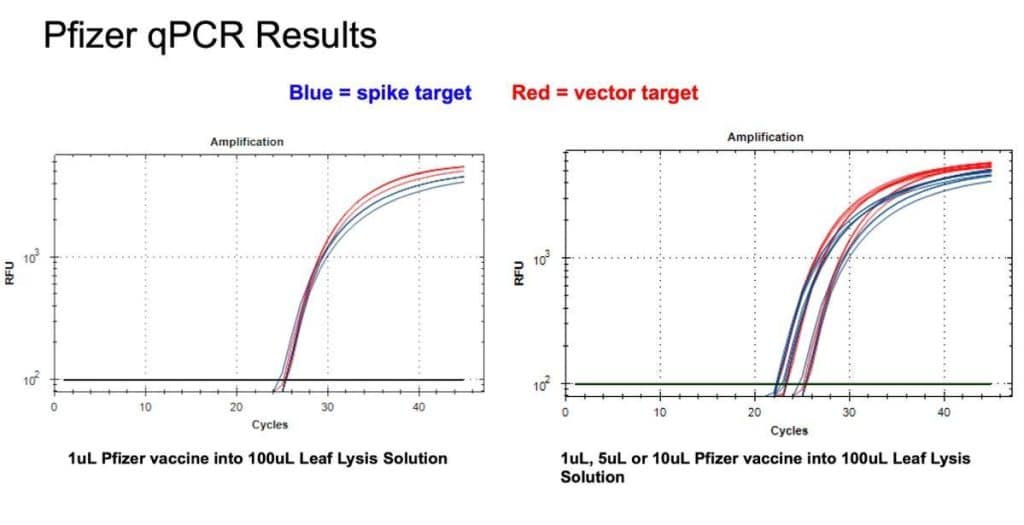
Figure 9. 1µl of the Pfizer bivalent vaccine placed in 100µl Leaf Lysis buffer for an 8 minute boil step delivers a CT of 24 for both Vector and Spike targets in qPCR (Left). Assay is responsive to 1,5,10µl of input (Right).
Figure 10. 1µl of the Pfizer bivalent vaccine placed in 100µl Leaf Lysis buffer for an 8 minute boil step delivers a CT of 20 and 12 for both Vector and Spike targets in RT-qPCR (Left). Assay is responsive to 1,5,10µl of input (Right)
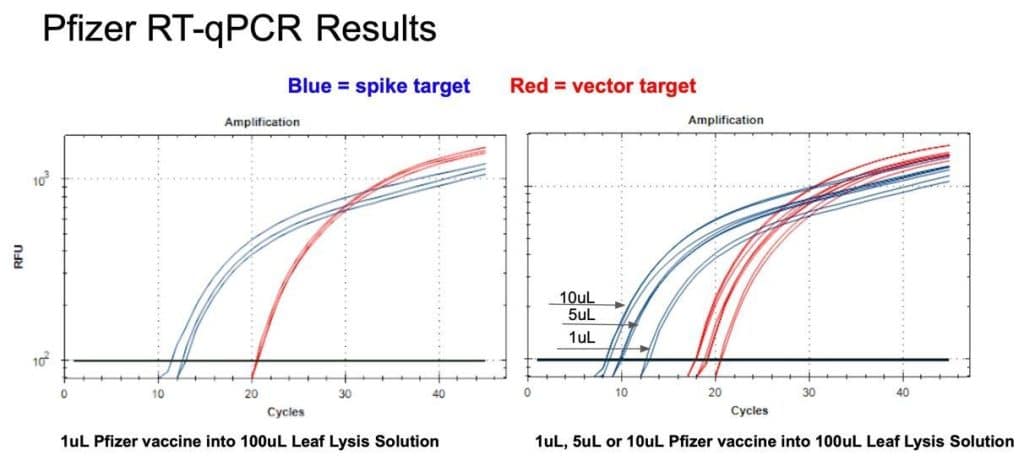
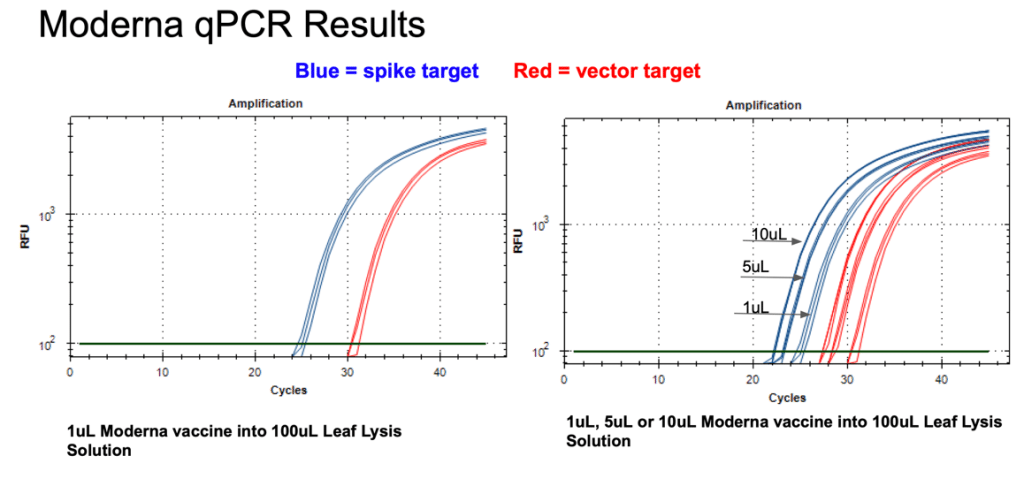
Figure 11. 1µl of the Moderna bivalent vaccine exhibits different CTs values for the spike and the vector targets (Left) with qPCR. This needs to be explored further as the assays provide equal CT scores on Pfizers’ vaccines and the sequence of the amplicon is identical between the two vector origins. There are 2 mismatches in the spike amplicons between Moderna and Pfizer but none of the mismatches are under a primer or probe. The assay is responsive to 1,5,10µl of direct boil mRNA (Right).
Figure 12. 1µl of the Moderna bivalent vaccine exhibits different CTs values for the spike and the vector targets (Left) with RT-qPCR. The large 10 CT shift between Spike and Vector needs to take into consideration that qPCR control shows a 5 CT offset. The boil preps can tolerate 1-10µl of vaccine (Middle and Right).
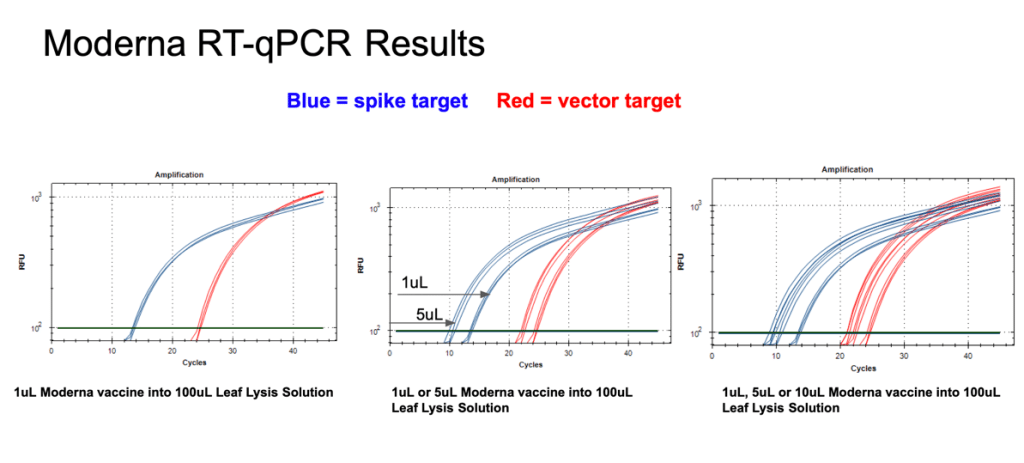
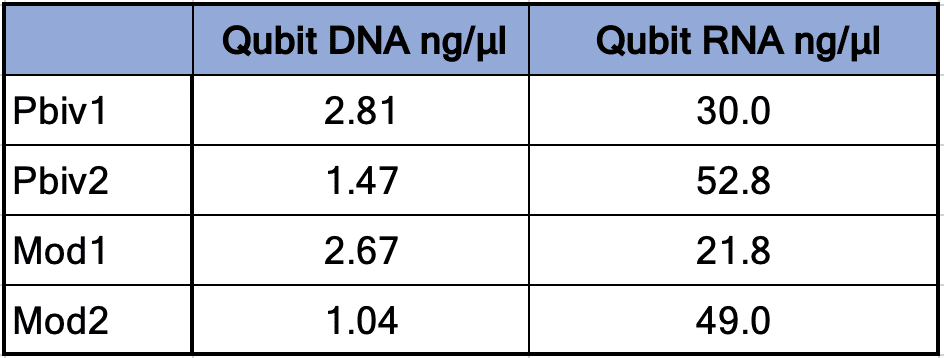
Table 1. Qubit™ 3 Fluorometry estimates 1.04-2.8 ng/µl of dsDNA in the vaccines and 21.8ng-52.8ng/µl of RNA.
Synthetic templates were synthesized with IDT to build RT-qPCR standard curves to benchmark CTs to the mass of DNA in the reaction. This method uses ideal templates and fails to quantitate DNA molecules smaller than the amplicon size. As expected, this method delivers lower DNA concentration estimates than Qubit™ 3 fluorometry or Agilent Tape Station™. It also represents an ideal environment which doesn’t capture the inhibition or primer depletion that can occur when large quantities of mRNA with identical sequence to your DNA target are co-present in a qPCR assay.
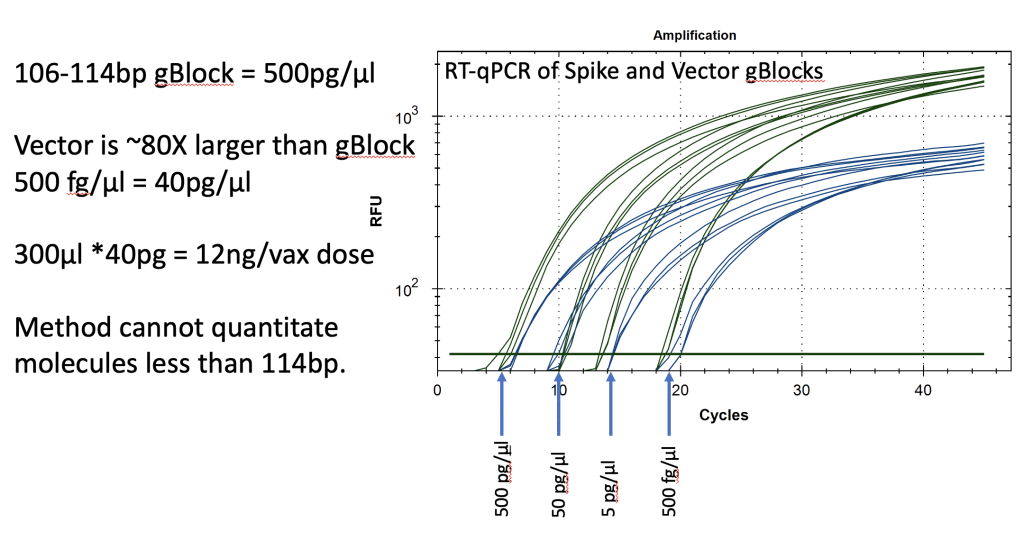
Figure 13. Two gBlocks were synthesized at IDT for Spike and Ori positive control templates used in an RT-qPCR assays. 10-fold serial dilutions were run in triplicate to correlate CT scores with picograms of DNA. The threshold is lowered from 102 for review of the background. CT of ~20 = 500fg/RT-qPCR reaction. Since 100bp targets only represent 1/80th of the vector DNA present as a potential contaminant, 500 fg/µl manifests in 40pg/µl of vector DNA. Any DNA that is DNase I treated and is smaller than the amplicon size cannot amplify or be quantitated with this method. This method will under quantitate DNase I treated samples compared to Qubit™ 3 or Agilent Tape Station™.
This work was further validated by testing 8 unopened Pfizer monovalent vaccines with both qPCR and RT-qPCR.
Figure 14. Moderna and Pfizer Bivalent vaccines were unopened and not expired (Top). 8 Monovalent Pfizer mRNA vaccines. These were unopened but past expiration (Bottom).
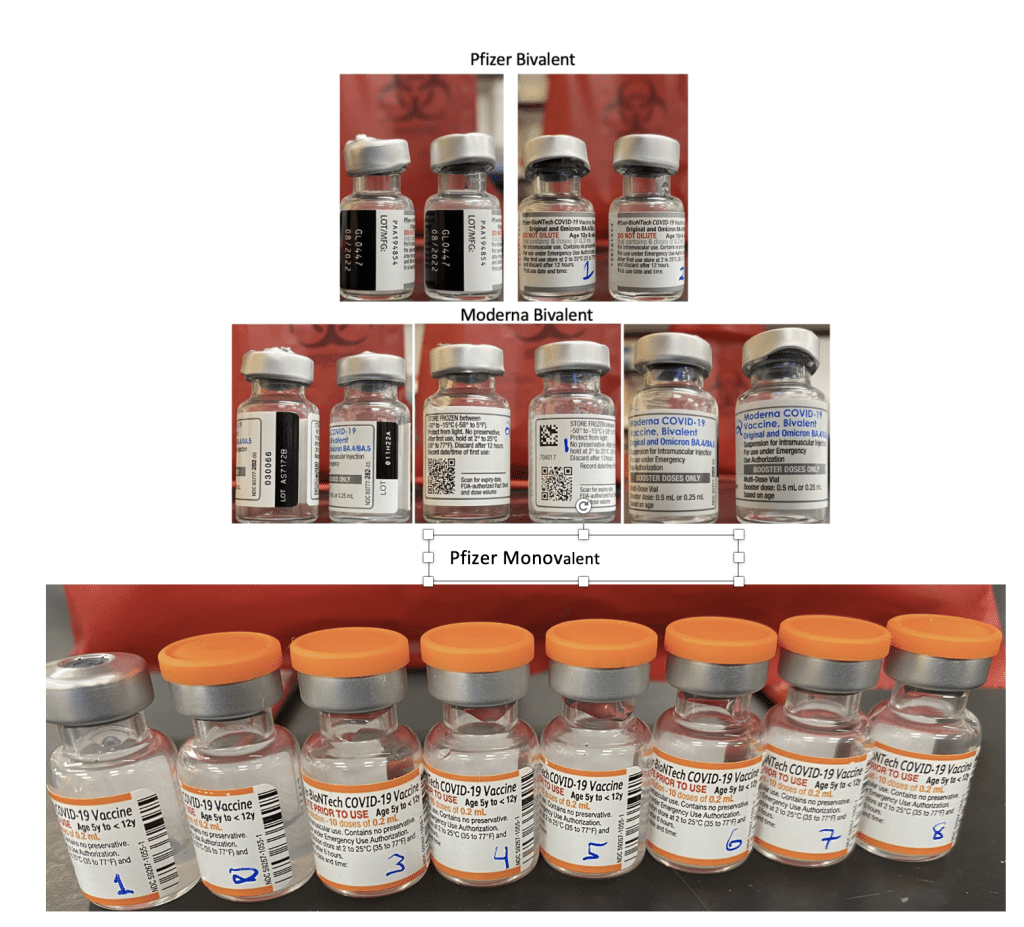
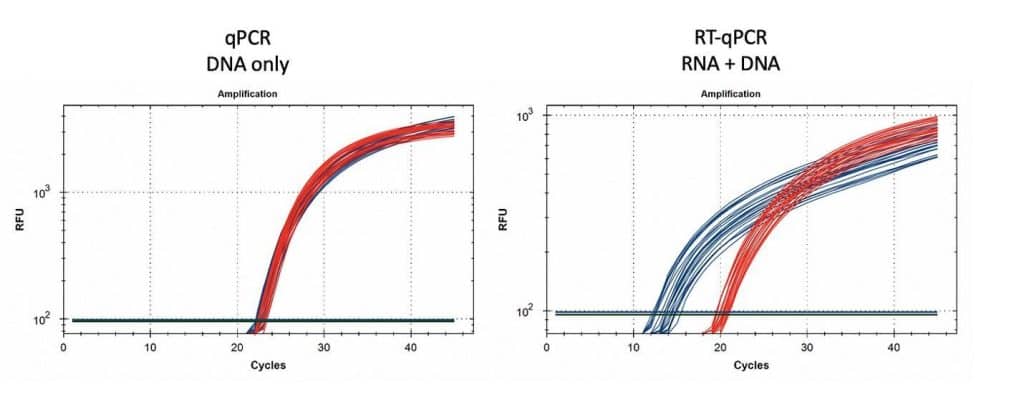
Figure 15. 1µl of vaccine boiled in 100µl of Leaf Lysis buffer was subjected to qPCR (left) and RT-qPCR (right) for Vector (red) and Spike (blue). 8 samples were tested in triplicate.
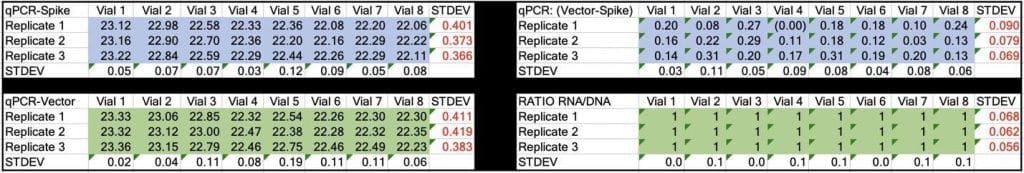
Table 2. CT values for Spike and Vector during qPCR (DNA only). Standard deviation for the triplicate measurements run horizontally in black font. Standard deviation for vial to vial run vertically in Red. Delta CT or (Vector CT minus Spike CT) represents the ratio of Spike to Vector DNA and should = 1
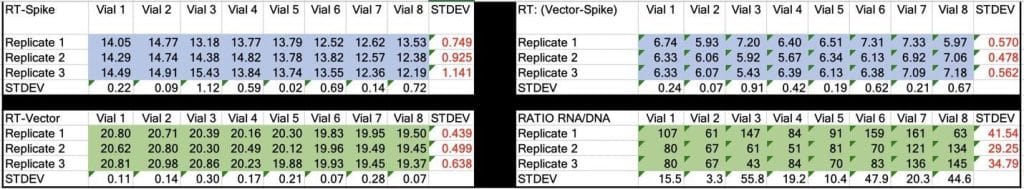
Table 3. CT values for Spike and Vector during RT-qPCR (RNA+DNA). Ratio of RNA:DNA ranges from 43:1 To 161:1. EMA allowable limit is 3030:1. This is 18-70 fold over the EMA limit.
Discussion
Multiple methods highlight high levels of DNA contamination in the both the monovalent and bivalent vaccines. While the Qubit™ 3 and Agilent Tape Station™ differ on their absolute quantification, both methods demonstrate it is orders of magnitude higher than the EMAs limit of 330ng DNA/ 1mg RNA. qPCR and RT-qPCR confirms the relative RNA to DNA ratio. An 11-12 CT offset should be seen between Spike and Vector RT-qPCR signals to represent a 1:3030 contamination limit (2^11.6 = 3100). Instead, we observe much smaller CT offsets (5-7 CTs) when looking at qPCR and RT-qPCR data with these vaccines. It should be noted that Qubit™ 3 and Agilent methods stain all DNA in solution while qPCR measures only amplifiable molecules without DNase I cut sites between the primers. The further apart you space the qPCR primers, the fewer Qubit™ 3 and Agilent detectable molecules will amplify. The primers used in this study are 106bp and 114bp apart, thus any molecules that are DNase I cut below this length will be undercounted wwith the qPCR methods relative to more general dsDNA measurements from Qubit™ 3 or Agilent Tape Station™.
This also implies that qPCR standard curves using 100% intact synthetic DNA standards will amplify more efficiently and thus undercount the total digested DNA contamination. For example, standard curves with 106-114bp synthetic templates provide CTs under 20 in the picogram range (not low nanogram range) suggesting large portions of the library are smaller than the minimum amplifiable size. Pure standards also do not contain high concentrations of modified mRNA with identical sequence which could serve as a competitive primer sink or inhibitor to qPCR methods.
Alternatively, the Qubit™ 3 and the Agilent Tape Station™ could be inflating the DNA quantification due to intercalating dye cross talk with N1-methylpseudouridine RNA. For this reason, we believe the ratio we observed when these molecules are more scrupulously interrogated with polymerases specific for each template type in qPCR and RT-qPCR is a more relevant metric. The EMA metric is also stated as such a ratio.
This also brings into focus if these EMA limits took into consideration the nature of the DNA contaminants. Replication competent DNA should arguably have a more stringent limit. DNA with mammalian promoters or antibiotic resistance genes may also be of more concern than just random background E.coli genomic DNA from a plasmid preparation (Sheng-Fowler et al. 2009). Background E.coli DNA was measured with qPCR and had CT over 35.
There has been a healthy debate about the capacity for SARs-CoV-2 to integrate into the human genome(Zhang et al. 2021). This work has inspired questions regarding the capacity for the mRNA vaccines to also genome integrate. Such an event would require LINE-1 driven reverse transcription of the mRNA into DNA as described by Alden et al. (Alden et al. 2022). dsDNA contamination of sequence encoding the spike protein wouldn’t require LINE-1 for Reverse Transcription and the presence of an SV40 nuclear targeting signal in Pfizer’s vaccine vector would further increase the odds of integration. This work does not present evidence of genome integration but does underscore that LINE-1 activity is not required given the dsDNA levels in these vaccines. The nuclear localization of these vectors should also be verified.
Prior sequencing of the monovalent vaccines from Jeong et al. only published the consensus sequence (Dae-Eun Jeong 2021). The raw reads for this project are not available and should be scrutinized for the presence of vector sequence.
Given these vaccines exceed the EMA limits (330ng/mg DNA/RNA) with the Qubit™ 3 and Agilent data and these data also exceed the FDA limit (10ng/dose) with the more conservative qPCR standard curves, we should revisit the lipopolysaccharide (LPS) levels. Plasmid contamination from E.coli preps are often co-contaminated with LPS. Endotoxins contamination can lead to anaphylaxis upon injection (Zheng et al. 2021).
A limitation of this study is the unknown provenance of the vaccine vials under study. These vials were sent to us anonymously in the mail without cold packs. RNA is known to degrade faster than DNA and it is possible poor storage could result in faster degradation of RNA than DNA. RNA as a molecule is very stable but in the presence of metals and heat or background ubiquitous RNases, it can degrade very quickly. All of the monovalent vaccines in this study are past the expiration date listed on the vial suggesting more work is required to understand the DNA to RNA ratios in fresh lots. The bivalent vaccines were not expired. The expiration dates for various vaccine lots have been continually extended by the manufacturers and used in patients. The publication of these qPCR primers may assist in surveying additional lots with more controlled supply chains. Studies evaluating vaccine longevity in breast milk or plasma may benefit from vector DNA surveillance as this sequence is unique to the vaccine and may persist longer than mRNA.
While the sequencing delivered full coverage of the plasmid backbones, it is customary to assemble plasmids from DNase I fragmented libraries. These methods have not discerned the ratio of linear versus circular DNA in the vials. While plasmid DNA is more competent and stable, linear DNA may have higher genome integration risks.
The intercalating dyes used in the Qubit™ 3 and Agilent systems are known to have low fluorescent cross talk with DNA and RNA but it is unknown to what degree N1-methylpseudouridine alters the specificity of these intercalating dyes. As a result, we have relied on the CT offsets between RT-qPCR and qPCR with the vector and spike sequence as the best relative assessment of the EMA ratio-metric regulation. These qPCR and RT-qPCR reagents may be useful in tracking these contaminants in vaccines, blood banks or patient tissues in the future
Methods
Purifying the mRNA from the LNPs
LiDs/SPRI purification
100µl of each vial was sampled (1/3rd to 1/5th of a dose)
- 5µl of 2% LiDs was added to 100µl of Vaccine to dissolve LNPs
- 100µl of 100% Isopropanol
- 233µl of Ampure (Beckman Genomics)
- 25µl of 25mM MgCl2 (New England Biolabs)
Samples were tip mixed 10X and incubated for 5 minutes for magnetic bead binding. Magnetic Beads were separated on a 96-well magnet plate for 10 minutes and washed twice with 200µl of 80% EtOH. The beads were left to air dry for 3 minutes and eluted in 100µl of ddH20. 2µl of eluted sample was run on an Agilent Tape Station™.
CTAB/Chloroform/SPRI purification of Vaccines
Some variability in qPCR performance was noted with our LiDs/SPRI purification method of the vaccines. This left some samples opaque and may represent residual LNPs in the purification. A CTAB/Chloroform/SPRI isolation was optimized to address this and used for further qPCR and Agilent electrophoresis. Briefly, 300µl of Vaccine was added to 500µl of CTAB (MGC solution A in SenSATIVAx MIP purification kit. #420004). The sample was then vortexed and heated for 5 minutes at 37°C. 800µl of chloroform was added, vortexed and spun at 19,000 rpms for 3 minutes. The top 250µl of aqueous phase was collected and added to 250µl of solution B and 1ml of magnetic binding buffer. Samples were vortexed and incubated for 5 minutes and magnetically separated. The supernatant was removed and the beads washed with 70% Ethanol two times. Samples were finally eluted in 300µl of MGC elution buffer.
Simple boil preparation for evaluating vaccine qPCR.
This boil prep process simply takes 1-10µl of the vaccine and dilutes it into a PCR compatible leaf lysis buffer and heats it (Medicinal Genomics part number 420208).
- 65°C for 6 minutes
- 95°C for 2 minutes
Library Construction for Sequencing
- 50µl of each 100µl sample was converted into RNA-Seq libraries for Illumina sequencing using the NEB NEBNext UltraII Directional RNA library Kit for Illumina (NEB#E7760S).
To enrich for longer insert libraries the fragmentation time was reduced from 15 minutes to 10 minutes and the First strand synthesis time was extended at 42°C to 50 minutes per the long insert recommendations in the protocol.
No Ribo depletion or PolyA enrichment was performed as to provide the most unbiased assessment of all fragments in the library. The library was amplified for 16 cycles according to the manufacturers protocol. A directional library construction method was used to evaluate the single stranded nature of the mRNA. This is an important quality metric in the EMA and TGA disclosure documents as dsRNA (>0.5%) can induce an innate immune response. dsRNA content is often estimated using an ELISA. Directional DNA sequencing offers a more comprehensive method for its estimation and was previously measured and 99.99% in Jeong et al. It is unclear how this may vary lot to lot or within the new manufacturing process for the newer bivalent vaccines.
RNase A treatment of the Vaccines
RNase A cleaves both uracils and cytosines. N1-methylpseudouridine is known to be RNAse-L resistant but RNase A will cleave cytosines which still exist in the mRNAs. This leaves predominantly DNA for sequencing. Vaccine mRNA that was previously sequenced and discussed here, was treated at 37°C for 30 minutes with 10µl of 20 Units/µl Monarch RNase A from NEB. The RNase reaction was purified using 1.5X of SenSATIVAx (Medicinal Genomics #420001). Sample were eluted in 20µl ddH20 after DNA purification. 15µl was used for DNA sequencing.
DNase treatment of the vaccines
50µl of CTAB purified vaccine was treated at 37°C for 30 minutes with 2µl DNase I and 6µl of DNase I buffer (Grim reefer MGC#420143). 2.5µl of LiDs Lysis buffer was added to stop the DNase reaction. Reactions were purified using 60µl 100% Isopropanol, 140µl Ampure, 15µl MgCl2. Magnetic beads were tip mixed 10 times, left for 5 minutes to incubate, magnetically separated and then washed twice with 80% EtOH.
Whole genome shotgun of RNase’d Vaccines.
15µl of the DNA was converted into sequence ready libraries using Watchmakers Genomics WGS library construction kit. This kit further fragments the DNA to smaller sizes making fragment length in the vaccines difficult to predict.
Qubit™ 3 Fluorometry
Qubit™ 3 fluorometry was performed using Biotum AccuBlue RNA Broad Range kit (#31073) and Biotum AccuGreen High Sensitivity dsDNA Quantitation Kit (#31066) according to the manufacturers instructions.
E.coli qPCR
Medicinal Genomics PathoSEEK™ E.coli Detection assay (#420102) was utilized according to the manufacturers instructions.
qPCR and RT-qPCR Spike Assay
- MedGen-Moderna_Pfizer_Janssen_Vax-Spike_Forward
- >AGATGGCCTACCGGTTCA
- MedGen-Moderna_Pfizer_Janssen_Vax-Spike_Reverse
- >TCAGGCTGTCCTGGATCTT
- MedGen-Moderna_Pfizer_Janssen_Vax-Spike_Probe
- >/56-FAM/CGAGAACCA/ZEN/
- GAAGCTGATCGCCAA/3IABkFQ/
- qPCR and RT-qPCR Vector Origin Assay
- MedGen_Vax-vector_Ori_Forward
- >CTACATACCTCGCTCTGCTAATC
- MedGen_Vax-vector_Ori_Reverse
- GCGCCTTATCCGGTAACTATC
- MedGen_Vax-vector_Ori_Probe
- /5HEX/AAGACACGA/ZEN/
- CTTATCGCCACTGGC/3IABkFQ/
Elute primer to 100uM according to IDT instructions.
Make 50X primer-probe mix.
- 25µl 100uM Forward Primer
- 25µl 100uM Reverse Primer
- 12.5µl 100uM Probe
- 37.5µl nuclease free ddH20.
Use 15µl of this mixture in the qPCR master mix setup seen below. (0.5µl primer/probe per reaction)
Use 10µl of this mixture in the RT-qPCR master mix setup seen below
Medicinal Genomics Master Mix kits used
Medical Genomics qPCR Master Kit
Medical Genomics pathoseek Master Kit
Reaction setup for 30 reactions of qPCR
- 114µl Enzyme Mix (green tube)
- 24µl Reaction Buffer (blue tube)
- 246µl nuclease free ddH20
- 15µl of Primer-Probe set Spike
- 15µl of Primer-Probe set Ori
Use 13.8µl of above Master Mix and 5µl of purified sample (1µl Vax DNA/RNA + 4µl ddH20 if CT <15)
Reaction setup for 34 reactions of RT-qPCR
- 200µl Enzyme mix
- 96µl nuclease free ddH20
- 20µl RNase Inhibitor (purple tube)
- 4µl DTT (green tube)
- 10µl Primer-Probe set Spike
- 10µl Primer-Probe set Ori
10µl of Master Mix and 1µl of Vax DNA/RNA
Medicinal Genomics MIP DNA Purification Kit used
he CTAB/Chloroform/SPRI based DNA/RNA isolation methods are described above.
Cycling conditions
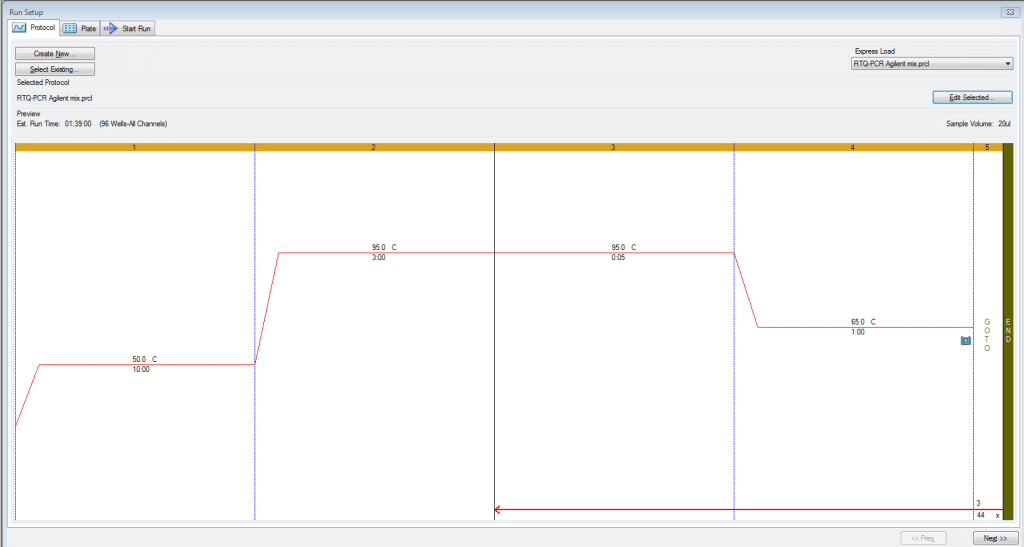
These conditions work for both qPCR and RT-qPCR. Note: The 50°C RT step can be skipped with qPCR. The MGC qPCR MasterMix kits used have a hot start enzyme which are unaffected by this 50°C step. For the sake of controlling RNA to DNA comparisons, we have put qPCR and RT-qPCR assays on the same plate and run the below program with the RT step included for all samples. Cycling Conditions used for qPCR and RT-qPCR
Sequences of amplicons for gBlock Positive Controls. Ori = 106bp, Spike = 114bp.
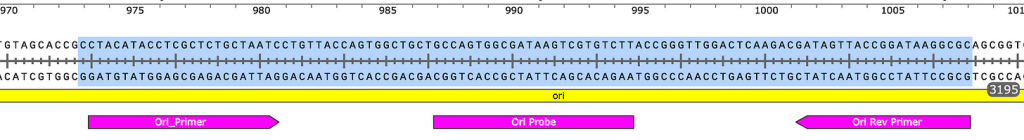
Ori target
Spike target
Sequencing Data
Raw Illumina Reads RNA-seq
Pfizer Bivalent Vial 1 Forwards Reads
- Pfizer Bivalent Vial 1 Reverse Reads
- Pfizer Bivalent Vial 2 Forwards Reads
- Pfizer Bivalent Vial 2 Reverse Reads
- Moderna Vial 1 Forwards Reads
- Moderna Vial 1 Reverse Reads
- Moderna Vial 2 Forward Reads
- Moderna Vial 2 Reverse Reads
Read files are run through sha256 (Hash and stash) and etched onto the DASH blockchain. The sha256 hash of the read file is spent into the OP_RETURN of an immutable ledger. If the hash of the file doesn’t match the hash in these transactions, the file has been tampered with.
- Pfizer Vial 1 Forward hash
- Pfizer Vial 1 Reverse hash
- Pfizer Vial 2 Forward hash
- Pfizer Vial 2 Reverse hash
- Moderna Vial 1 Forward hash
- Moderna Vial 1 Reverse hash
- Moderna Vial 2 Forward hash
- Moderna Vial 2 Reverse hash
Megahit Assemblies
Illumina Reads mapped back to Megahit Assemblies
- Pfizer Vial 1 BAM File. Index File
- Pfizer Vial 2 BAM File. Index File
- Moderna Vial 1 BAM File. Index File
- Moderna Vial 2 BAM File. Index File
Q30 Filtered Illumina Reads (use these for transcriptional error rate estimates)
FastQ-Filter download: usage> fastq-filter -e 0.001 -o output.fastq input.fastq
- Pfizer bivalent Vial 1 Forward Reads
- Pfizer bivalent Vial 1 Reverse Reads
- Pfizer bivalent Vial 2 Forward Reads
- Pfizer bivalent Vial 2 Reverse Reads
- Moderna bivalent Vial 1 Forward Reads
- Moderna bivalent Vial 1 Reverse Reads
- Moderna bivalent Vial 2 Forward Reads
- Moderna bivalent Vial 2 Reverse Reads
Q30 BAM files. Q30 Reads mapped against Megahit assemblies
- Pfizer Vial 1 q30-BAM file. Index File
- Pfizer Vial 2 q30-BAM file. Index File
- Moderna Vial 1 q30-BAM file. Index File
- Moderna Vial 2 q30-BAM file. Index File
IGVtools error by base on q30 reads
Fields = Position in contig, Positive stand (+)A, +C, +G, +T, +N, +Deletion, +Insertion, Negative strand -A, -C, -G, -T, -N, -Deletion, -Insertion
Analysis pipeline
Reads were demultiplexed and processed with:
- Trimgalore – Removes Illumina Sequencing adaptors.
- Megahit- assembles reads into contigs.
- Megahit for SARs-CoV-2
- Samtools- generates BAM files for viewing in IGV.
- Samtools stats used to calculate outie reads.
- BWA-mem- Short read mapper used to align reads back to the assembled references.
- SnapGene software- (www.snapgene.com)- Used to visualize and annotate expression vectors
- IGV– Integrated Genome Viewer used to visualize Illumina sequencing reads.
RNase Treated Libraries-BAM files
contig specific BAM files were created using samtools samtools view -h input.bam contig_name -O BAM > contig.bam; samtools index contig.bam; Samtools stats run on a each contig in each assembly. for out_prefix in `ls *.sort.bam | perl -pe “s/.sort.bam//”`; do mkdir -p ${out_prefix}-samtools-stats; for contig in `samtools view -H ${out_prefix}.sort.bam | grep “^@SQ” | cut -f 2 | perl -pe “s/SN\://”`; do echo “Now calculating stats for ${contig}/$out_prefix…”; samtools stats ${out_prefix}.sort.bam $contig > ${out_prefix}-samtools-stats/${contig}-samtools-stats.txt; done; done
Kevin McKernan, Yvonne Helbert, Liam T. Kane, Stephen McLaughlin